

The Electron Thermal Conductivity of Pu and Zr Substituted γ-U
Nuclear Science and Engineering / Volume 200 / Number 7 / July 2026 / Pages 1688-1709
Research Article / dx.doi.org/10.1080/00295639.2025.2537479

Articles are hosted by Taylor and Francis Online.
Uranium alloys are attractive recycled nuclear fuels because of their high thermal conductivity (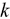 ) and fissile density.
) and fissile density. 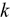
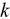 .
. 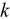 ,
,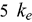 ) of U-Pu-Zr compositions are calculated using density functional theory. The electronic structure is evaluated to understand the effects of plutonium (Pu) and zirconium (Zr) substitution on the
) of U-Pu-Zr compositions are calculated using density functional theory. The electronic structure is evaluated to understand the effects of plutonium (Pu) and zirconium (Zr) substitution on the 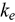 of
of 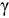 -U. Alloys of up to 37.5 at. % Pu and 37.5 at. % Zr are examined. Two methods are applied to calculate
-U. Alloys of up to 37.5 at. % Pu and 37.5 at. % Zr are examined. Two methods are applied to calculate 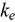 ; we find that the accuracy of each method depends on the electronic and mass similarities between the solute and solvent atoms. Specifically, when the solute atom is similar in electronic structure and mass, the more accurate method is that which employs the electron relaxation time of
; we find that the accuracy of each method depends on the electronic and mass similarities between the solute and solvent atoms. Specifically, when the solute atom is similar in electronic structure and mass, the more accurate method is that which employs the electron relaxation time of 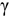 -U, while if the elements are dissimilar, a mixed method that mixes several parameters associated with
-U, while if the elements are dissimilar, a mixed method that mixes several parameters associated with  from each element in the alloy is best. The introduction of all alloying elements decreases
from each element in the alloy is best. The introduction of all alloying elements decreases  ; however, in binary compounds, Pu and Zr have different effects. Pu flattens the electronic bands but compensates for this deleterious effect by increasing electron density near the Fermi level. Zr flattens the electronic bands more severely without adding electron density near the Fermi level. Therefore, Zr decreases
; however, in binary compounds, Pu and Zr have different effects. Pu flattens the electronic bands but compensates for this deleterious effect by increasing electron density near the Fermi level. Zr flattens the electronic bands more severely without adding electron density near the Fermi level. Therefore, Zr decreases 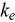 more than Pu in binary compounds. In ternary compounds, the difference between Pu and Zr is minimal due to the phononic change from the large mass change of Zr substitution, even at 12.5 at. %. Thus, we predict that higher loadings of Pu, and potentially other actinides, can be added to U-Pu-Zr compositions for faster recycling of spent fuel without sacrificing
more than Pu in binary compounds. In ternary compounds, the difference between Pu and Zr is minimal due to the phononic change from the large mass change of Zr substitution, even at 12.5 at. %. Thus, we predict that higher loadings of Pu, and potentially other actinides, can be added to U-Pu-Zr compositions for faster recycling of spent fuel without sacrificing 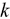 . We also note that these
. We also note that these 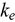 calculation methods can be applied to non-fuel alloys that require
calculation methods can be applied to non-fuel alloys that require 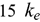 predictions, such as cladding, heat exchanger, and structural materials.
predictions, such as cladding, heat exchanger, and structural materials.